Methods
Cell passaging and cell culture
Neuroblastoma cell culture and retinoic acid treatment
Before the treatment could begin, the cells were passaged from the SK-N-SH cell line and grown in a 10cm dish for the experiments this paper encompasses. This plate begins in a sterile hood with 1mL of neuroblastoma cells and 9mL of media stock which is a 1:1 ratio of Dulbecco’s Modified Eagle Media (DMEM) and F-12 with 10% fetal bovine serum with pen strap. This media provides nutrients for the cells to grow and also contain substances that help to prevent contamination. These cells were then incubated in a climate of 37 degrees Celsius with 5% CO2. These cells are incubated until the plate confluence is roughly 95%-100%. After the desired confluence is reached the 10cm dish into four 2cm dished. Two of these dishes were controls and two were treated with retinoic acid.
During the passaging process the old media was removed and the cells are washed with 10mL of PBS which removes dead cells and other unnecessary waste in the dish. Then, 3mL of Trypsin was added which breaks down a protein in the cell that allows it to adhere to the dish. The Trypsin was removed and the plate was then incubated for five minutes to allow the Trypsin to breakdown the protein. After five minutes were over the plate was removed from the incubator and returned to the hood. There, 10mL of the media stock was replaced. The media was pipetted around the dish multiple times in order to collect all of the cells. The cells were then passaged into the 2cm dishes containing cover slips in quantities of .4mL per dish to reach a ~20% confluence in the dishes . Then 1.6mL of the media stock was added to each of the plates to bring the total volume in the four plates to 2mL. Next, two of the plates received 4 microliters of retinoic acid which was dissolved in ethanol and DMSO. The two control dishes were treated with ethanol and DMSO to make everything consistent about the two sets of plates besides the retinoic acid treatment. These cells then had the media periodically changed and the treatment of retinoic acid and the control will be replaced in the media for the course of 5 days.
Cell fixation and staining for microscopy
Once the duration of the experiment ended the cells will be fixed with formaldehyde after being washed 3 times with PBS. The formaldehyde solution contains 200 microliters of 37% dilution of formaldehyde and 1.8mL of PBS. Each slide receives 1mL of the solution then, after these cells were fixed they were then placed in PBS and frozen down in liquid nitrogen until they were ready to be stained.
Phalloidin Stain
The first stain, phalloidin, begun by washing the fixed cells 3 times with PBS wash. In order to allow the dye to attach to its respective region within the cell, the membrane must become permeable. To do this .25mL of Triton X-100 is mixed with 9.75mL of PBS. Then, 2mL of the diluted solution is added to each of the plates. Next, the plates are placed on an orbiting instrument for 10 minutes. Once the time is over the liquid is removed and the cells are again washed 3 times with PBS. To block nonspecific regions from the phalloidin stain the cells receive a solution of BSA Blocking Buffer and PBS. 4 microliters of BSA Blocking Buffer is added to 396 microliters of PBS. The plates are then treated with 200 microliters of the solution. The plates are then returned to the orbiting instrument for another 10 minutes. Once the times has finished the liquid is removed the cells are washed once with PBS. After, 5 microliters of phalloidin is added to 195 microliters of BSA. Since phalloidin is light sensitive these steps require aluminum foil around the dishes and mixture to protect from deterioration. 100 microliters of the solution is then added to each plate and returned to the orbiting instrument for 20 minutes. The cells are then washed a final time with PBS and the coverslips are removed from the dishes and a dye DAPI is placed on the cover slide to stain the nucleus of the cell. The cover slips are then placed on slides and sealed on two ends. The cells were then observed under a confocal microscope and images were collected using microscopy.
GFAP and β-Tubulin Stain
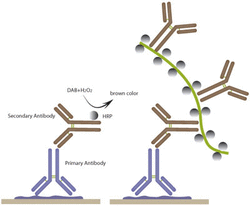
Another set of cells were stained with GFAP and β-Tubulin specific antibodies and then treated with immunoflourecent dyes in order to be seen under a confocal microscope. First, the cells were treated with a non-specific blocking buffer and a solution to permeate the cell membrane. This solution composed of 30 microliters of Triton X-100, 10 microliters of goat serum, and 1.96mL of PBS. Each slide received 1mL of the solution and sat for [X] minutes. Then, the cells were washed in PBS and treated with the antibodies. The antibodies were in an antibody dilution buffer composed of 180 microliters of BSA, 27 microliters of Triton X-100, and 1.593 mL of PBS. Then 4 microliters of GFAP and 4 microliters of β-Tubulin antibodies were added to 800 microliters of the antibody dilution buffer. Each slide received 404 microliters of the solution. The solution incubated at room temperature on the plate for an hour. Then, the solution was removed from the plate and the slides were washed with PBS. Later, a secondary antibody stain is added which binds to the antibody and gives a fluorescent characteristic to the cells when under a confocal microscope. These antibodies were incubated at room temperature for an hour, after they are removed and the slides were washed in PBS. The slides were then removed from the slides are placed on a cover slide that has DAPI on it to stain the nucleus. These cells are then viewed under a confocal microscope that uses frequencies of light to produce a fluorescence where the dye has bonded on the cell. This allows for viewing of the morphological characteristics of a cell and the ability to see other specific proteins that are stained. Images are then collected from the confocal microscope for further analysis. This picture is an example of immunoflourescent staining. The primary antibody attaches itself to the desired protein like a key to a lock. Then a secondary antibody stain similarly attaches itself to the primary antibody stain. This secondary stain has florescent properties and can be seen under a confocal microscope.
Methylene Blue Stain
The final stain was the methylene blue stain which is used to help observe morphological changes in the cells. First, the cells are fixed and washed with PBS rinse. Then the cells are treated with 300 microliters of methylene blue for 15 minutes. The cells are then washed 3 times for a duration of 5 minutes each. PBS rinse is placed in the dish and is used to keep the cells from drying out. Finally, the cells are viewed under a light microscope in order to see the stained cells. Pictures were then recorded for data.
Conductivity Test
The conductivity test was used to determine the active potential of the treated and untreated cells and was also used to determine the ability of the cell’s sodium ion pumps. Cells were grown up specifically for conductivity testing. The cells were grown on cover slides inside of 2cm petri dishes. These cover slides were then placed in a rig that measures electrophysiology and is able to send electrical signals through the cell. The cell was latched onto by a pipette tip that is connected to the cell membrane. Then the cell was tested for active potential and for sodium spike amplitude which are bother characteristic of neurons. The data was then recorded based on the amount of energy that is able to be conducted.
